
Protective effects of cyclosporine A and hypothermia on neuronal mitochondria in a rat asphyxial cardiac arrest model
a b s t r a c t
Background: Cyclosporine A (CsA) was neuroprotective in the settings of traumatic brain injury and stroke. We sought to investigate the protective effects of CsA and hypothermia on neuronal mitochondria after cardiac arrest. Methods and Results: Five groups were included: sham (S), normothermia (N), CsA (C), hypothermia (H), and CsA plus hypothermia (C + H). Cardiac arrest was induced by 10 min of asphyxia. CsA (10 mg/kg) was administered im- mediately after return of spontaneous circulation in the CsA groups. Temperature of the rats was maintained at 33 +- 0.5 ?C after return of spontaneous circulation in the hypothermia groups. Hippocampal mitochondria were measured after 2 h of resuscitation. Mitochondrial transmembrane potential was significantly higher in the C, the H, and the C + H groups than in the N group and was higher in the C + H group than in the C and the H groups. Cytosolic Cytochrome c was significantly higher in the N group. superoxide dismutase activity was significantly lower in the N group than in the other groups and was higher in the C and the C + H groups than in the H group. Malondialdehyde concentration was significantly higher in the N group.
Conclusions: CsA or hypothermia used immediately after resuscitation enhanced mitochondrial transmembrane po- tential, kept cytochrome c from releasing out of the mitochondria, increased Superoxide dismutase activity, and de- creased malondialdehyde concentration in hippocampus. Moreover, the protective effects of CsA were reinforced by hypothermia. One of the mechanisms that hypothermia protected neuronal mitochondria from damage was inhibiting the opening of mitochondrial permeability transition pore.
(C) 2016
Introduction
The high morbidity and mortality associated with cardiac arrest re- mains a difficult problem. Though many cardiac arrest victims are success- fully resuscitated initially, only a small number of them survive to hospital discharge. Post-resuscitation syndrome is an important factor that affects survival after resuscitation. It is a complex state characterized by myocar- dial dysfunction, brain injury, global Ischemia-reperfusion injury and systemic inflammatory response [1]. Most patients die during the post-resuscitation period due to neuronal damage, which develops as a consequence of global cerebral ischemia during cardiac arrest [2]. Further- more, as many as 40% to 50% of the surviving patients suffer from perma- nent cognitive impairment [3,4]. Therefore, improving the long-term health of cardiac arrest patients and optimizing the treatment of post- resuscitation syndrome are required.
? Sources of support: Funding was obtained from the Nature Science Foundation, De- partment of Science and Technology, Heilongjiang, China (Grant #: LC2012C40).
* Corresponding author at: Department of Anesthesiology, the Third Affiliated Hospital, Harbin Medical University, Heilongjiang, China 150081.
E-mail address: [email protected] (F. Han).
The energy imbalance due to Mitochondrial dysfunction leads to neu- ronal damage. The main mitochondrial damage after resuscitation is the opening of mitochondrial permeability transition pore (mPTP). mPTP, a non-specific channel, allows molecules as large as 1500 Da to enter the mitochondrial matrix, as well as water and other substances, resulting in mitochondrial swelling, cytochrome c release into the cytosol, activa- tion of the apoptotic cascade, loss of mitochondrial transmembrane po- tential (??m), ion balance disruption, oxidative phosphorylation uncoupling, increased Reactive oxygen species production, and re- duced energy generation. Robust bursts of ROS further induce mPTP opening, creating a vicious cycle [5-7]. Cyclosporine A (CsA), an inhibitor of mPTP, inhibits activation of mPTP and pore formation by inhibiting matrix CypD interaction with pore proteins [8,9]. CsA demonstrated a significant increase in ??m, accompanied by lower levels of intramitochondrial Ca2+ and reactive oxygen species production in trau- matic brain injury [10]. The neuroprotective properties of CsA were medi- ated through modulation of the mPTP and maintenance of mitochondria homeostasis [10]. CsA administration reduced infarct size, DNA fragmen- tation and apoptotic bodies, and inflammatory responses in a stroke model in the rat brain [11]. CsA treatment at onset of resuscitation de- creased the mitochondrial swelling of the heart and improved both post-cardiac arrest myocardial dysfunction and survival [12]. The
http://dx.doi.org/10.1016/j.ajem.2016.02.066
0735-6757/(C) 2016
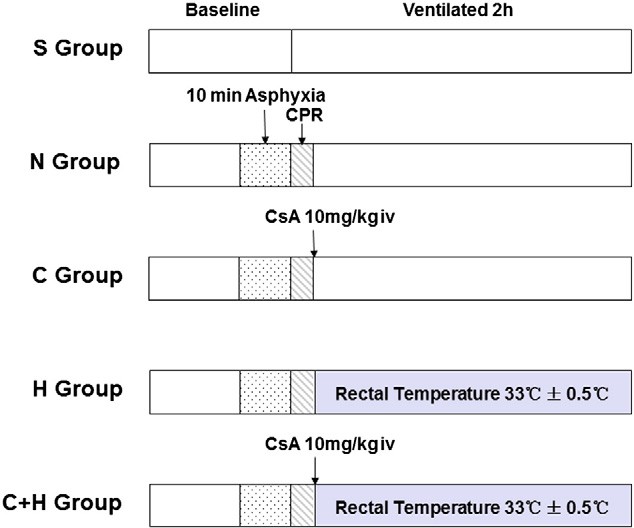
Fig. 1. The experimental protocol. S group, Sham group; N group, normothermia group; C group, CsA group (10 mg/kg, i.v.); H group, Mild hypothermia group; C + H group, CsA + mild hypothermia group.
mitochondrial integrity and respiration function of the heart are amelio- rated under CsA treatment at onset of resuscitation [12]. These reports in- dicate CsA might be a promising agent in treating neuronal mitochondrial dysfunction after cardiac arrest [10-12]. Therapeutic mild hypothermia is a well-documented method that has been proven effective during cere- bral resuscitation by many studies [13-21]. However, few studies have attempted to determine what the influence of CsA and hypothermia on the opening of neuronal mPTP and the protection of mitochondrial func- tion following cardiac arrest. In this study, we sought to find out the pro- tective effects exerted by CsA and mild hypothermia on neuronal mitochondria after resuscitation in a rat asphyxial cardiac arrest model.
Materials and methods
The experimental protocol for the study was approved by the Insti- tutional Animal Care and Use Committee of The Third Affiliated Hospi- tal, Harbin Medical University. Male Wistar rats (260-310 g) purchased from the Changchun City Billion Adams Laboratory Animal Technology Co., Ltd. were allowed to acclimate for 6 days before begin- ning the experiment.
Experimental groups
The rats were divided into five groups (n = 12 each, Fig. 1): sham group (S group); normothermia group (N group), in which the rats were subjected to cardiac arrest induced by 10 min of asphyxia, and the rectal temperature was maintained at 37 +- 0.5 ?C after the return of spon- taneous circulation (ROSC); CsA group (C group), in which CsA (10 mg/kg) [12,22] was administered intravenously immediately
following ROSC in the setting of normothermia; mild hypothermia group (H group), in which rectal temperatures were maintained at 33 +- 0.5 ?C following ROSC; CsA plus mild hypothermia group (C + H group), in which CsA was administered intravenously in combination with mild hypothermia. Six animals in the each group were decapitated to determine ??m of the hippocampus after 2 h of resuscitation. The re- maining six animals in the each group were also decapitated for the mea- surement of the expression of cytoplasmic cytochrome c and caspase-3, the activity of Superoxide dismutase , and the concentration of Malondialdehyde of the hippocampus after 2 h of resuscitation.
Animal preparation
The rats were anesthetized with 4% isoflurane. Mechanical ventila- tion was initiated using a volume rodent ventilator (model 683, Harvard Apparatus, South Natick, MA, USA) at the rate of 40/min, I:E = 1:1, FiO2 = 0.5. The tidal volume (8-12 ml/kg) was regulated by end-tidal CO2 maintained between 35 and 45 mmHg. During surgical preparation, the rectal temperature of the rats was maintained between 37.0 +-
0.5 ?C. Anesthesia was sustained with 2.5% isoflurane.
Asphyxia-induced cardiac arrest
After balancing to maintain normal mean arterial pressure (MAP), heart rate (HR), and blood gas values, the experimental animals were given vecuronium (2 mg/kg, intravenously) 5 min before asphyxia. Car- diac arrest was induced via asphyxia by turning off the ventilator and clamping the endotracheal tube. Cardiac arrest was confirmed by a MAP b 20 mmHg. The rectal temperature was maintained at 37 +-
0.5 ?C during the entire asphyxia period.
Resuscitation and ventilation
Following 10 min of asphyxia, chest compressions were performed at a rate of 200~ 300/min in combination with 0.01 mg/kg epinephrine and 2 mEq/kg NaHCO3. Ventilation was resumed at the same time. Body temperature was maintained according to different groups via either heating or Surface cooling following ROSC. The experimental animals were ventilated for 2 h and then hippocampus was isolated.
Isolation of hippocampal mitochondria
Hippocampal mitochondria were extracted using a tissue mitochon- dria isolation kit (Beyotime Institute of Biotechnology, China). The ho- mogenate of hippocampus was centrifuged at 1000 g for 5 min at 4 ?C to remove nuclei and any unbroken cells. The supernatant was collected and centrifuged at 35,000 g for 10 min at 4 ?C to obtain the mitochondri- al fraction (deposition).
The characteristics of the each group at baseline.
|
S group |
N group |
C group |
H group |
C + H group |
|
|
Weight (g) |
280.0 +- 15.6 |
290.3 +- 16.4 |
279.3 +- 15.3 |
292.9 +- 12.9 |
277.7 +- 14.2 |
|
MAP (mmHg) |
98.3 +- 7.2 |
102.2 +- 11.6 |
99.8 +- 11.1 |
101.3 +- 11.7 |
92.1 +- 5.5 |
|
HR (bpm) |
321.7 +- 20.0 |
330.2 +- 8.6 |
333.1 +- 13.0 |
321.5 +- 15.2 |
335.8 +- 14.5 |
|
Temp (?C) |
36.9 +- 0.3 |
36. 9 +- 0.2 |
37.0 +- 0.4 |
37.1 +- 0.3 |
37.0 +- 0.4 |
|
pH |
7.39 +- 0.05 |
7.39 +- 0.05 |
7.39 +- 0.04 |
7.38 +- 0.04 |
7.36 +- 0.02 |
|
PaCO2 (mmHg) |
41.9 +- 7.1 |
41.5 +- 8.0 |
46.5 +- 9.5 |
42.8 +- 6.7 |
41.0 +- 8.8 |
|
PaO2 (mmHg) |
248.2 +- 98.7 |
276.0 +- 95.1 |
320.4 +- 56. 5 |
290.2 +- 93.5 |
225.9 +- 77.7 |
|
Na+ (mmol/L) |
135.3 +- 10.1 |
138.3 +- 8.3 |
140.4 +- 5.0 |
146.0 +- 10.3 |
136.3 +- 11.2 |
|
K+ (mmol/L) |
4.3 +- 1.1 |
3.8 +- 0.6 |
3.9 +- 0.7 |
3.7 +- 0.4 |
3.6 +- 0.6 |
|
Cl? (mmol/L) |
110.3 +- 10.8 |
114.8 +- 7.3 |
112.6 +- 5.1 |
117.7 +- 7.4 |
109.5 +- 12.2 |
MAP, mean arterial pressure; HR, heart rate; Temp, temperature.
Mitochondrial transmembrane potential assay
??m of the hippocampal mitochondria was evaluated using both an FACS Calibur flow cytometer (BD Bioscience, San Jose, CA) and a sensi- tive fluorescent JC-1 probe (Mitochondrial membrane potential detec- tion kit, Beyotime Institute of Biotechnology, China). ??m was determined using the ratio of JC-1 aggregates (red) to JC-1 monomers (green). Mitochondrial depolarization was expressed as the decrease in the intensity ratio of red/green fluorescence.
Cytoplasmic cytochrome c and caspase-3 analysis via Western blotting
Equal amounts of protein (100 ug) were loaded. The primary antibod- ies and concentrations used were as follows: cytochrome c (1:2000, Cell Signaling Technology, Beverly, MA, USA), cleaved caspase-3 (1:1000, Cell Signaling Technology, Beverly, MA, USA) and ?-actin (1:1000, Beijing Zhongshan Golden Bridge Biotechnology Co. Ltd., Beijing, China). The im- munoblots were next processed with the appropriate secondary antibod- ies (1:5000, Beijing Zhongshan Golden Bridge Biotechnology Co. Ltd., Beijing, China) for 1 h at Room temperature.
Measurement of SOD activity and MDA concentration
SOD activity was measured using a SOD assay kit (Jiancheng Bioen- gineering Institute, Nanjing, China). The chromogenic reaction mixture was added to equal amounts of protein in the each sample at room tem- perature for 10 min. The absorbance of the mixture was then measured spectrophotometrically at 550 nm. SOD activity was expressed as units/ mg of tissue protein. The level of lipid peroxidation products (MDA) was measured using an MDA assay kit (Jiancheng Bioengineering Institute, Nanjing, China). Equal amounts of protein from the each sample were mixed with a chromogenic reagent at 95 ?C for 40 min. The samples were centrifuged at 4000 rpm for 10 min at 4 ?C. The absorbance of the supernatants was measured spectrophotometrically at 532 nm. The MDA level was calculated in millimicromoles per milligram of pro- tein based on a standard curve.
Statistical analysis
All values are presented as means +- standard errors of the mean. The difference between groups was evaluated using One-way ANOVA, followed by the Newman-Keuls multiple comparison test. Values of P b .05 were considered statistically significant. All statistics were assessed using SPSS 17.0 (SPSS Inc., Chicago, IL, USA).
Results
There were no differences in body weight, MAP, HR, blood gas values, and temperature among the groups at baseline (Table 1). The time of cardiac arrest and the time of cardiopulmonary resuscitation were no difference among the groups (Table 2). MAP after resuscitation was no difference among the groups (Fig. 2). Rectal temperature of dif- ferent groups was controlled based on the experimental protocol.
Hippocampal mitochondrial transmembrane potential
??m, measured by JC-1 staining, is presented in Fig. 3. The fluores- cence intensity ratios of the aggregates and monomers in the H (0.96 +-
CA time and CPR time of the each group.
N group C group H group C + H group
|
CA time (sec) 194.1 +- 15.0 |
194.6 +- 18.3 |
191.3 +- 11.0 |
187.4 +- 17.9 |
|
CPR time (sec) 71.3 +- 59.2 |
61.9 +- 28.3 |
55.6 +- 24. 5 |
103.3 +- 95.7 |
CA, cardiac arrest; CPR, cardiopulmonary resuscitation.
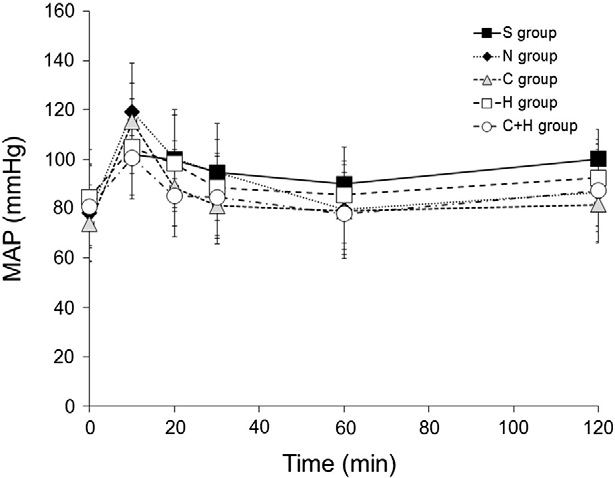
Fig. 2. Mean arterial pressure (MAP) after return of spontaneous circulation.
0.12), the C (1.01 +- 0.12) and the C + H (1.18 +- 0.08) groups were sig- nificantly higher than in the N group (0.68 +- 0.04, P b .05). The fluores- cence intensity ratio of the aggregates and monomers in the C + H group was higher than in the H and the C groups (P b .05) and was higher in the S group (1.46 +- 0.03) than in the other groups (P b .05).
Cytoplasmic expression of cytochrome c and caspase-3
The average level of cytosol cytochrome c among the different groups was determined using band densitometry of western blotting (Fig. 4). Cytochrome c was significantly higher in the N, the C, the H, and the C + H groups than in the S group (P b .05). Cytochrome c was significantly higher in the N group than in the C, the H, and the C + H groups (P b .05). There were no differences among the H, the C, and the C + H groups. No caspase-3 expression was noted in any of the groups (data not shown).
SOD activity and MDA concentration
SOD activity was significantly lower in the N group than in the other groups (P b .05, Fig. 5). SOD activity was higher in the C and the C + H groups than in the H group (P b .05). MDA content was highest in the N group com- pared with the other groups (P b .05, Fig. 6). MDA concentration was not sig- nificantly different among the S, the C, the H, and the C + H groups.
Discussion
CsA inhibits mPTP opening, attenuates acute mitochondrial dysfunc- tion, and exerts Neuroprotective effects after traumatic brain injury and stroke [10,11]. Few studies determine what the influence of CsA on neu- ronal mitochondria function following cardiac arrest. To our knowledge, this study is the first time to indicate that CsA used immediately follow- ing ROSC after cardiac arrest enhances ??m, keeps cytochrome c from releasing out of the mitochondria, increases SOD activity, and decreases MDA concentration in hippocampus. Moreover, the administration of CsA in combination with hypothermia results in greater protective ef- fects. Our results also demonstrate hypothermia protected neuronal mi- tochondria was via inhibiting the opening of mPTP.
mPTP, a nonspecific pore, has been implicated in the ischemic brain injury and the initiation of apoptosis [23,24]. Many factors (such as intra-cellular calcium overload, reactive oxygen species bursts, and re- duced energy generation) induce the mPTP opening which results in decreased ??m during I/R injury [25]. A decrease of ??m is considered a sign of early apoptosis [26]. The molecular structure of mPTP is not well known. However, it has been suggested that the voltage- dependent anion carrier (VDAC) in the outer membrane, the adenine
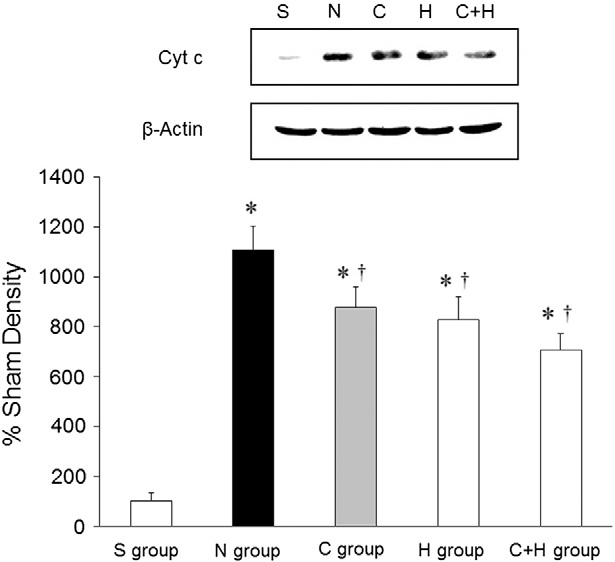
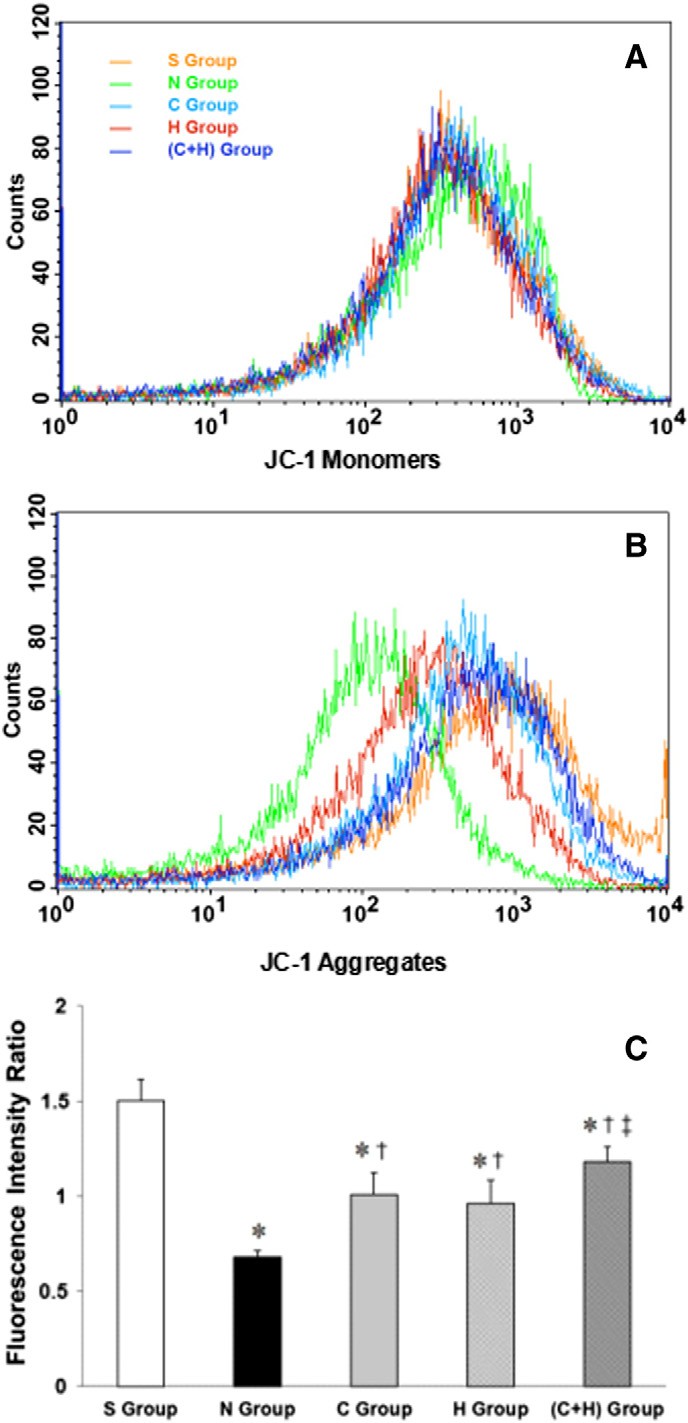 Fig. 3. The intensities of the JC-1 monomers and aggregates, respectively (A, B). The inten- sity ratio of the aggregates and monomers (C). * P b .05, vs. the S group; + P b .05, vs. the N group; ? P b .05, vs. the C group and the H group.
Fig. 3. The intensities of the JC-1 monomers and aggregates, respectively (A, B). The inten- sity ratio of the aggregates and monomers (C). * P b .05, vs. the S group; + P b .05, vs. the N group; ? P b .05, vs. the C group and the H group.
nucleotide translocase (ANT) in the inner mitochondrial membrane, and cyclophilin D (Cyp-D) in the matrix are involved in the composition of the mPTP [27]. CsA, a specific inhibitor of mPTP permeability, exerts protective effects after I/R injury, as reported in a variety of in vitro [28,29] and in vivo [30] animal models. CsA prevents CyP-D from bind- ing to adenine-nucleotide translocator (ANT). This binding antagonizes mPTP formation and desensitizes the mPTP to Ca2+, which interrupts both the apoptotic and necrotic cascades [31-33]. Cyp-D deficient mice exhibit reduced brain infarct sizes following acute Middle cerebral artery occlusion and reperfusion [23,34,35]. In addition to inhibiting the mPTP, CsA also inhibits calcineurin, which might alter intracellular cal- cium homeostasis after I/R injury. Although some immunosuppressants
Fig. 4. Representative cytosolic cytochrome c expression. * P b .05, vs. the S group; + P b .05, vs. the N group.
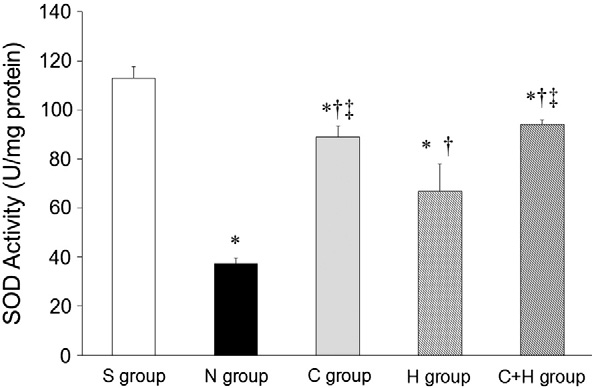
Fig. 5. SOD activity. * P b .05, vs. the S group; + P b .05, vs. the N group; ? P b .05, vs. the H group.
(such as FK506) inhibit calcineurin, they do not inhibit the mPTP and therefore do not prevent mitochondrial dysfunction [36,37]. Uchino et al. demonstrated that CsA completely suppresses mitochondrial swelling and release, whereas FK506 exhibits only partial inhibitory ef- fects, indicating that CsA is more effective as a neuroprotective agent in the setting of ischemia than FK506 [38]. These findings suggest that the protective effects exerted by CsA on mitochondrial function are primar- ily caused by mPTP inhibition rather than calcineurin inhibition.
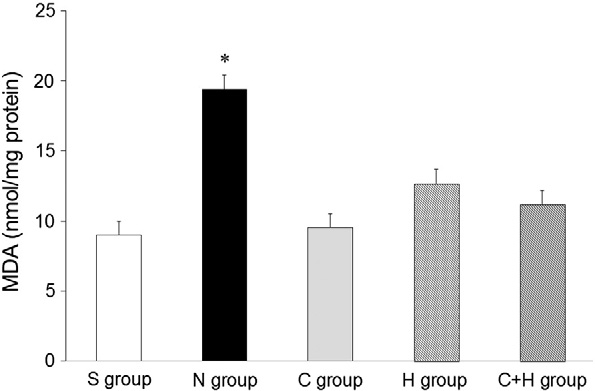
Fig. 6. MDA concentration. * P b .05, vs. the other groups.
Utilizing gerbil ischemia models, another study demonstrates that CsA exerts greater neuroprotective effects on insulted brains than FK506 [39]. These protective effects were demonstrated via the inhibition of cytochrome c release. Cytochrome c releasing into the cytosol occurs primarily via mPTP opening [6,7,39]. The results of our study are consis- tent with these findings. Either CsA or mild hypothermia used after re- suscitation decreases the amount of cytochrome c release from neuronal mitochondria. It suggests the inhibition of mPTP opening by CsA and hypothermia.
CsA exerts protective effects after I/R injury on the brain, the heart, and the liver which were demonstrated by many reports [8,40-44]. However, the poor ability of CsA to cross the blood-brain barrier (BBB) limits its neuroprotective effects. Several studies have demon- strated that CsA exerts neuroprotective effects when it is already pres- ent in the neuron as a result of its pre-administration, as well as during cases in which the BBB is disrupted prior to neuronal injury [10,11,45]. The results of this study demonstrate that CsA enhances ??m, decreases the releasing of cytochrome c, improves the activity of SOD, and decreases MDA concentration. These indicate a temporary increasing of BBB permeability following cardiac arrest induces more CsA crossing the BBB to achieve neuroprotective effects.
Therapeutic hypothermia is the promising method that has been proven effective in cerebral resuscitation by many reports [13-21]. Hy- pothermia induced neuroprotection is associated with inhibiting the opening of mPTP, reducing release of pro-apoptotic substances from mi- tochondria, and increasing mitochondrial respiration in a swine model of cardiac arrest [46]. Free radical and Ca2+ overload are two major reg- ulators of mPTP. The exact mechanism by which hypothermia inhibits mPTP opening was not investigated. Inhibition of ischemia-induced cal- cium overload could be involved [47]. Interestingly, the results of our study indicated ??m, cytochrome c released out of the mitochondria, and MDA concentration in hippocampus after resuscitation were in the same level between the C and the H groups. Hypothermia showed the same protective effects as CsA when used after cardiac arrest. It sug- gested one of the mechanisms of hypothermia to protect neuronal mito- chondria from damage might be the same as CsA by inhibiting the opening of mPTP which was consist with the previous report [46,47]. Moreover, in our study, hypothermia reinforced the protective effects of CsA. However, therapeutic hypothermia increases Infection risk by suppression of the host’s response to infection, creating limitations in clinicians’ ability to promptly detect and diagnose infection, requiring interventions such as invasive catheters and mechanical ventilators which breach natural barriers and also impairs wound healing by trig- gering cutaneous vasoconstriction [48]. CsA has immunosuppressive ac- tivity. Inhibition of calcineurin by CsA occurs via binding to the immunophilin cyclophilin, which prevents the dephosphorylation of NFAT and its subsequent translocation from the cytoplasm to the nucle- us in an IL-2-mediated process. Inhibition at this level thereby prevents activation of promoters of T-cell activation and overall immune re- sponse [49]. Although no reports showed additive effects of CsA with hypothermia on infection, increased risk should be considered.
The release of cytochrome c from the mitochondria into the cytosol
activates the intrinsic apoptotic pathway via apoptosome formation. No caspase-3 activity was noted in any of the groups following 2 h of reper- fusion in our study. A possible reason is that the increased expression of caspase-3 occurred at 6-8 h following ischemia in the hippocampus, peaked at 24 h after the reperfusion, and lasted until 72 h following is- chemia [50,51].
SOD catalyzes the dismutation of the superoxide anion to H2O2, which is subsequently reduced to H2O and O2 by peroxidases [52]. How- ever, during the reperfusion of ischemic tissues, these natural defenses can be overcome, resulting in the conversion of hydrogen peroxide to the hydroxyl radical, a short-lived type of ROS. A variety of biological molecules, including amino acids, membrane transport proteins, and nucleic acids, can sustain damage due to hydroxyl radical exposure [53,54]. Because neurons exhibit low antioxidative enzyme activity,
they are vulnerable to ROS during I/R injury [55]. In this study, both CsA and mild hypothermia increased SOD activity and decreased MDA content, which facilitated ROS elimination and protect neurons from the damage of free radical.
There were limitations in this study. First, the end point of this ex- periment was two hours after resuscitation. The endpoint was deter- mined according to previous report [56]. In the future studies, more prolonged observation periods, neurological behavior evaluation, and survival are necessary to be investigated. Second, this study did not en- tail observing the side effects of CsA. In addition to the immunosuppres- sive affects, CsA has toxic effects. CsA forms a high affinity complex with cyclophilin-A which inhibits the calcium-activated protein phospha- tase, calcineurin. Nephrotoxicity, hypertension, hirsuitism, gingival hy- poplasia, elevated transaminases and bilirubin, and neurotoxicity (such as seizures) are adverse side effects observed in receiving CsA treatment. However, sanglifehrin A, another potent inhibitor of the mPTP, is not effective against calcineurin. The protective effects on neu- ronal mitochondria after resuscitation exerted by Sanglifehrin A should be investigated in the future studies. Third, we did not compare differ- ent dosages of CsA used after resuscitation. The optimal dosage of CsA for the protection of neuronal mitochondria after cardiac arrest should be investigated.
In conclusion, CsA or hypothermia used immediately after resuscita- tion enhanced ??m, kept cytochrome c from releasing out of the mito- chondria, increased SOD activity, and decreased MDA concentration in hippocampus. Moreover, the protective effects of CsA were reinforced by hypothermia. One of the mechanisms that hypothermia protected neuronal mitochondria from damage was inhibiting the opening of mPTP.
Acknowledgments
This study was supported by the Nature Science Foundation, Department of Science and Technology, Heilongjiang, China (Grant #: LC2012C40).
References
- Neumar RW, Nolan JP, Adrie C, Aibiki M, Berg RA, Bottiger BW, et al. Post-cardiac ar- rest syndrome: epidemiology, pathophysiology, treatment, and prognostication. A consensus statement from the International Liaison Committee on Resuscitation (American Heart Association, Australian and New Zealand Council on Resuscitation, European Resuscitation Council, Heart and Stroke Foundation of Canada, InterAmerican Heart Foundation, Resuscitation Council of Asia, and the Resuscita- tion Council of Southern Africa); the American Heart Association Emergency Cardio- vascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; and the Stroke Council. Circulation 2008;118(23):2452-83.
- Laver S, Farrow C, Turner D, Nolan J. Mode of death after admission to an intensive care unit following cardiac arrest. Intensive Care Med 2004;30(11):2126-8.
- Lim C, Alexander MP, LaFleche G, Schnyer DM, Verfaellie M. The neurological and cognitive sequelae of cardiac arrest. Neurology 2004;63(10):1774-8.
- van Alem AP, de Vos R, Schmand B, Koster RW. Cognitive impairment in survivors of out-of-hospital cardiac arrest. Am Heart J 2004;148(3):416-21.
- Mazzeo AT, Beat A, Singh A, Bullock MR. The role of mitochondrial transition pore, and its modulation, in traumatic brain injury and delayed neurodegeneration after TBI. Exp Neurol 2009;218(2):363-70.
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J 1999;341(Pt 2):233-49.
- Halestrap AP. The mitochondrial permeability transition: its molecular mechanism and role in reperfusion injury. Biochem Soc Symp 1999;66:181-203.
- Nicolli A, Basso E, Petronilli V, Wenger RM, Bernardi P. Interactions of cyclophilin with the mitochondrial inner membrane and regulation of the permeability transi- tion pore, and cyclosporin A-sensitive channel. J Biol Chem 1996;271(4):2185-92.
- Lemasters JJ, Qian T, He L, Kim JS, Elmore SP, Cascio WE, et al. Role of mitochondrial inner membrane permeabilization in necrotic cell death, apoptosis, and autophagy. Antioxid Redox Signal 2002;4(5):769-81.
- Sullivan PG, Thompson MB, Scheff SW. Cyclosporin A attenuates acute mitochondri- al dysfunction following traumatic brain injury. Exp Neurol 1999;160(1):226-34.
- Leger PL, De Paulis D, Branco S, Bonnin P, Couture-Lepetit E, Baud O, et al. Evaluation of cyclosporine A in a stroke model in the immature rat brain. Exp Neurol 2011; 230(1):58-66.
- Huang CH, Tsai MS, Hsu CY, Su YJ, Wang TD, Chang WT, et al. Post-cardiac arrest myocardial dysfunction is improved with cyclosporine treatment at onset of resus- citation but not in the reperfusion phase. Resuscitation 2011;82(Suppl. 2):S41-7.
- Abella BS, Rhee JW, Huang KN, Vanden Hoek TL, Becker LB. induced hypothermia is underused after resuscitation from cardiac arrest: a current practice survey. Resusci- tation 2005;64(2):181-6.
- Merchant RM, Soar J, Skrifvars MB, Silfvast T, Edelson DP, Ahmad F, et al. Therapeutic hypothermia utilization among physicians after resuscitation from cardiac arrest. Crit Care Med 2006;34(7):1935-40.
- Hagerdal M, Harp J, Nilsson L, Siesjo BK. The effect of induced hypothermia upon ox- ygen consumption in the rat brain. J Neurochem 1975;24(2):311-6.
- Horiguchi T, Shimizu K, Ogino M, Suga S, Inamasu J, Kawase T. Postischemic hypo- thermia inhibits the generation of hydroxyl radical following transient forebrain is- chemia in rats. J Neurotrauma 2003;20(5):511-20.
- Berger C, Schabitz WR, Georgiadis D, Steiner T, Aschoff A, Schwab S. Effects of hypo- thermia on excitatory amino acids and metabolism in stroke patients: a microdialy- sis study. Stroke 2002;33(2):519-24.
- Hachimi-Idrissi S, Van Hemelrijck A, Michotte A, Smolders I, Sarre S, Ebinger G, et al. Postischemic mild hypothermia reduces neurotransmitter release and astroglial cell proliferation during reperfusion after Asphyxial cardiac arrest in rats. Brain Res 2004;1019(1-2):217-25.
- Xu L, Yenari MA, Steinberg GK, Giffard RG. Mild hypothermia reduces apoptosis of mouse neurons in vitro early in the cascade. J Cereb Blood Flow Metab 2002; 22(1):21-8.
- Siesjo BK, Bengtsson F, Grampp W, Theander S. Calcium, excitotoxins, and neuronal death in the brain. Ann N Y Acad Sci 1989;568:234-51.
- Busto R, Globus MY, Dietrich WD, Martinez E, Valdes I, Ginsberg MD. Effect of mild hypothermia on ischemia-induced release of neurotransmitters and free fatty acids in rat brain. Stroke 1989;20(7):904-10.
- Okonkwo DO, Melon DE, Pellicane AJ, Mutlu LK, Rubin DG, Stone JR, et al. Dose- response of cyclosporin A in attenuating traumatic Axonal injury in rat. Neuroreport 2003;14(3):463-6.
- Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, et al. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A 2005;102(34):12005-10.
- Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, et al. The mito- chondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta 1998;1366(1-2):177-96.
- Miura T, Tanno M. Mitochondria and GSK-3beta in cardioprotection against ische- mia/reperfusion injury. Cardiovasc Drugs Ther 2010;24(3):255-63.
- Adhihetty PJ, Ljubicic V, Menzies KJ, Hood DA. Differential susceptibility of subsarcolemmal and intermyofibrillar mitochondria to apoptotic stimuli. Am J Phys- iol Cell Physiol 2005;289(4):C994-C1001.
- Sims NR, Muyderman H. Mitochondria, oxidative metabolism and cell death in stroke. Biochim Biophys Acta 2010;1802(1):80-91.
- Akao M, O’Rourke B, Kusuoka H, Teshima Y, Jones SP, Marban E. Differential actions of cardioprotective agents on the mitochondrial death pathway. Circ Res 2003; 92(2):195-202.
- Halestrap AP, Connern CP, Griffiths EJ, Kerr PM. Cyclosporin A binding to mitochon- drial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol Cell Biochem 1997;174(1-2):167-72.
- Argaud L, Gateau-Roesch O, Muntean D, Chalabreysse L, Loufouat J, Robert D, et al. Specific inhibition of the mitochondrial permeability transition prevents lethal re- perfusion injury. J Mol Cell Cardiol 2005;38(2):367-74.
- Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some ne- crotic but not apoptotic cell death. Nature 2005;434(7033):652-8.
- Halestrap AP, Woodfield KY, Connern CP. Oxidative stress, thiol reagents, and mem- brane potential modulate the mitochondrial permeability transition by affecting nucle- otide binding to the adenine nucleotide translocase. J Biol Chem 1997;272(6):3346-54.
- Woodfield K, Ruck A, Brdiczka D, Halestrap AP. Direct demonstration of a specific inter- action between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem J 1998;336(Pt 2):287-90.
- Wang X, Carlsson Y, Basso E, Zhu C, Rousset CI, Rasola A, et al. Developmental shift of cyclophilin D contribution to hypoxic-ischemic brain injury. J Neurosci Off J Soc Neurosci 2009;29(8):2588-96.
- Wang X, Han W, Du X, Zhu C, Carlsson Y, Mallard C, et al. Neuroprotective effect of bax-inhibiting peptide on neonatal brain injury. Stroke 2010;41(9):2050-5.
- Griffiths EJ, Halestrap AP. Further evidence that cyclosporin A protects mitochondria from calcium overload by inhibiting a matrix peptidyl-prolyl cis-trans isomerase. implications for the immunosuppressive and toxic effects of cyclosporin. Biochem J 1991;274(Pt 2):611-4.
- Friberg H, Ferrand-Drake M, Bengtsson F, Halestrap AP, Wieloch T. Cyclosporin A, but not FK 506, protects mitochondria and neurons against hypoglycemic damage and implicates the mitochondrial permeability transition in cell death. J Neurosci 1998;18(14):5151-9.
- Uchino H, Minamikawa-Tachino R, Kristian T, Perkins G, Narazaki M, Siesjo BK, et al. Differential neuroprotection by cyclosporin A and FK506 following ischemia corre- sponds with differing abilities to inhibit calcineurin and the mitochondrial perme- ability transition. Neurobiol Dis 2002;10(3):219-33.
- Domanska-Janik K, Buzanska L, Dluzniewska J, Kozlowska H, Sarnowska A, Zablocka
B. Neuroprotection by cyclosporin A following transient brain ischemia correlates with the inhibition of the early efflux of cytochrome C to cytoplasm. Brain Res Mol Brain Res 2004;121(1-2):50-9.
He L, Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Lett 2002;512(1-3): 1-7.
- Griffiths EJ, Halestrap AP. Protection by cyclosporin A of ischemia/reperfusion- induced damage in isolated rat hearts. J Mol Cell Cardiol 1993;25(12):1461-9.
- Clarke SJ, McStay GP, Halestrap AP. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J Biol Chem 2002;277(38): 34793-9.
- Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the per- meability transition pore in mitochondria devoid of cyclophilin D. J Biol Chem 2005; 280(19):18558-61.
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005;434(7033):658-62.
- Okonkwo DO, Povlishock JT. An intrathecal bolus of cyclosporin A before injury pre- serves mitochondrial integrity and attenuates axonal disruption in traumatic brain injury. J Cereb Blood Flow Metab 1999;19(4):443-51.
- Gong P, Hua R, Zhang Y, Zhao H, Tang Z, Mei X, et al. Hypothermia-induced neuro- protection is associated with reduced mitochondrial membrane permeability in a swine model of cardiac arrest. J Cereb Blood Flow Metab 2013;33(6):928-34.
- Tissier R, Couvreur N, Ghaleh B, Bruneval P, Lidouren F, Morin D, et al. Rapid cooling preserves the ischaemic myocardium against mitochondrial damage and left ven- tricular dysfunction. Cardiovasc Res 2009;83(2):345-53.
- Kuchena A, Merkel MJ, Hutchens MP. Postcardiac arrest temperature management: infectious risks. Curr Opin Crit Care 2014;20(5):507-15.
- Garcia-Saenz-de-Sicilia M, Mukherjee S. The adverse pharmacology of calcineurin inhibitors and their impact on Hepatitis C recurrence after liver transplantation: im- plications for clinical practice. Expert Rev Clin Pharmacol 2012;5(5):587-93.
- Li J, Han B, Ma X, Qi S. The effects of propofol on hippocampal caspase-3 and bcl-2 expression following forebrain ischemia-reperfusion in rats. Brain Res 2010;1356: 11-23.
- Chen J, Nagayama T, Jin K, Stetler RA, Zhu RL, Graham SH, et al. Induction of caspase- 3-like protease may mediate delayed neuronal death in the hippocampus after tran- sient cerebral ischemia. J Neurosci 1998;18(13):4914-28.
- Ohta H, Adachi T, Hirano K. Internalization of human extracellular-superoxide dis- mutase by bovine aortic Endothelial cells. Free Radic Biol Med 1994;16(4):501-7.
- Anaya-Prado R, Toledo-Pereyra LH, Lentsch AB, Ward PA. Ischemia/reperfusion inju- ry. J Surg Res 2002;105(2):248-58.
- Halliwell B, Gutteridge JM, Cross CE. free radicals, antioxidants, and human disease: where are we now? J Lab Clin Med 1992;119(6):598-620.
- Chan PH. Role of oxidants in ischemic brain damage. Stroke 1996;27(6):1124-9.
- Hausenloy DJ, Duchen MR, Yellon DM. Inhibiting mitochondrial permeability transi- tion pore opening at reperfusion protects against ischaemia-reperfusion injury. Cardiovasc Res 2003;60(3):617-25.